Presented in Nature Communications, the technology is called RADICL-seq and enables comprehensive mapping of each RNA, captured while interacting with all the genomic regions that it targets, where many RNAs are likely to be important for genome regulation and structure maintenance.
It was previously believed that RNA functions mostly as an intermediary in building proteins based on a DNA template (fig. 1a), with very rare exceptions such as ribosomal RNAs. However, with the development of genomic analysis, it turned out that not all DNA regions encode RNA, and not all transcribed RNA encodes proteins.
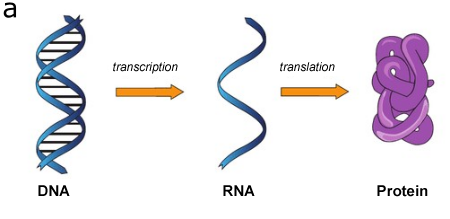
Figure 1. Gene expression (a): The DNA sequence of a gene is copied to make an RNA molecule (transcription), then the RNA sequence is decoded (translated) to build a protein molecule
Although the number of noncoding RNAs and those that encode proteins is about the same, the function of most noncoding RNA is still not entirely clear.
Every type of cell has its own set of active genes, resulting in the production of specific proteins. This makes a brain cell different from a blood cell of the same organism — despite both sharing the same DNA. Scientists are now coming to the conclusion that RNA is one of the factors that determine which genes are expressed, or active.
Long noncoding RNAs are known to interact with chromatin — DNA tightly packaged with proteins (fig. 1b). Chromatin has the ability to change its conformation, or “shape,” so that certain genes are either exposed for transcription or concealed. Long noncoding RNAs contribute to this conformation change and the resulting change in gene activity by interacting with certain chromatin regions. To understand the regulatory potential of RNA — in addition to it being a template for protein synthesis — it is important to know which chromatin region any given RNA interacts with.
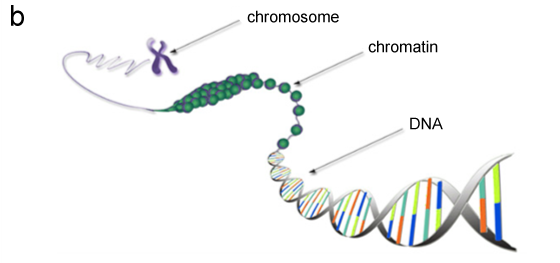
Figure 1. Gene expression (b) Inside a cell nucleus, a DNA molecule is packaged by special proteins in chromatin, which makes up a chromosome. Credit: Genetic Code Table, and Diagram of Chromatin from the Wiring Diagram Database
RNAs interact with chromatin inside the cell nucleus by binding to chromatin-associated proteins that fold a DNA molecule. There are several technologies that can map such RNA-chromatin interactions. However, all of them have significant limitations. They tend to miss interactions, or require a lot of input material, or disrupt the nuclear structure.
To address these shortcomings, a RIKEN-led team has presented a new method: RNA and DNA Interacting Complexes Ligated and Sequenced, or RADICL-seq for short. The technique produces more accurate results and keeps the cells intact up until the RNA-chromatin contacts are ligated.
The main idea of the RADICL-seq method is the following. First, the RNA is crosslinked to proteins located close to it in the nucleus of cells with formaldehyde. Then, DNA is cut into pieces by digesting it with a special protein. After that, the technology employs RNase H treatment to reduce ribosomal RNA content, thus increasing the accuracy of the result. Then, by using a bridge adapter (a molecule with single-stranded and double-stranded ends) the proximal DNA and RNA are ligated (fig. 2a). After the reversal of crosslinks, the RNA-adapter-DNA chimera is converted to double-stranded DNA for sequencing (fig. 2b), revealing the sequence of the ligated RNA and DNA.
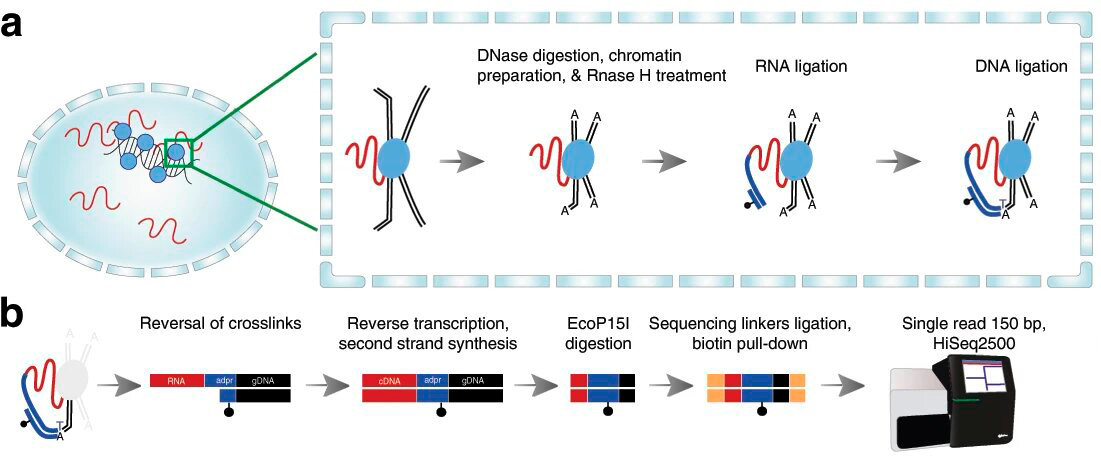
Figure 2. Reactions in the cell nucleus (a): The RNA is depicted in red, with the DNA shown as black double lines and a linking molecule in dark blue. The light blue circles stand for proteins. The black dot is a molecule that allows to single out the DNA-RNA hybrid in solution. Explanations are provided in the text. Reactions in solution (b): protein removal, second strand synthesis, clipping to predetermined size, addition of sequences used for recognition, and sequencing. Credit: Alessandro Bonetti et al./Nature Communications
In comparison with other existing methods, RADICL-seq mapped RNA-chromatin interactions with a higher accuracy. Moreover, the superior resolution of the technology allowed the team to detect chromatin interactions not only with the noncoding but also with the coding RNAs, including those found far from their transcription locus. The research confirmed that long noncoding RNAs play an important role in the regulation of gene expression occurring at a considerable distance from the regulated gene.
This technology can also be used to study cell type-specific RNA-chromatin interactions. The scientists proved it by looking at two noncoding RNAs in a mouse cell, one of them possibly associated with schizophrenia. They found that an interaction pattern between chromatin and those RNAs in two different cells — the embryonic stem cell and the oligodendrocyte progenitor cell — correlated with preferential gene expression in those cell types (fig. 3).
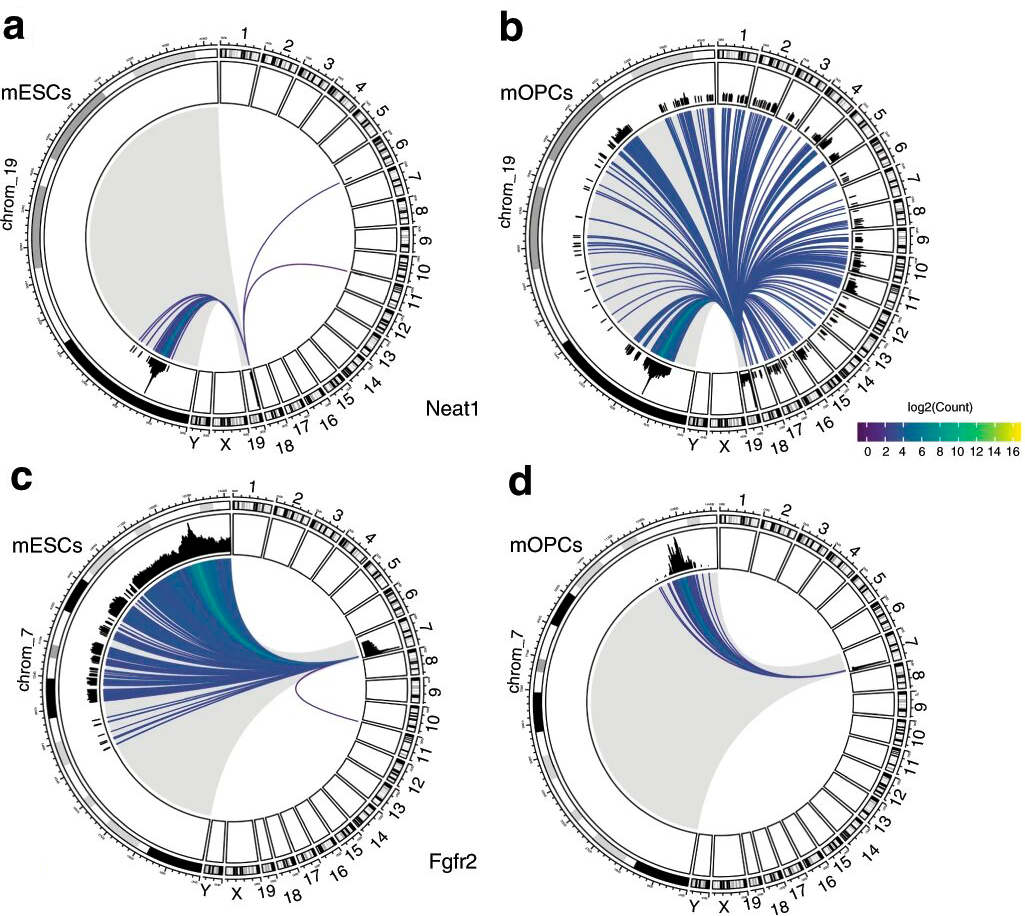
Figure 3. Noncoding RNA-chromatin interaction patterns: Neat1 (a, b) and Fgfr2 (c, d) in mouse embryonic stem cells (mESC) and mouse oligodendrocyte progenitor cells (mOPC). Neat1 and Fgfr2 come from chromosome Nos. 18 and 7, respectively. Credit: Alessandro Bonetti et al./Nature Communications
The new method’s flexibility means scientists can gather additional biological information by modifying the experiment. In particular, this technology can make it possible to identify direct RNA-DNA interactions not mediated by chromatin proteins. The analysis performed by bioinformaticians from the Research Center of Biotechnology and MIPT showed that not only the standard double helix interactions between DNA and RNA but also those involving RNA-DNA triplexes could participate in gene regulation. Also, such interactions highlight the significance of noncoding RNA in protein targeting to particular gene loci.
“We are planning to conduct further research on the role of RNA in the regulation of gene expression, chromatin remodeling, and ultimately, cell identity. Hopefully, we will be able to regulate genes by using these noncoding RNAs in the near future. This can be especially helpful for treating diseases,” says Yulia Medvedeva, who leads the Regulatory Transcriptomics and Epigenomics group at the Research Center of Biotechnology, RAS, and heads the Lab of Bioinformatics for Cell Technologies at MIPT. She also manages the grant project supported by the Russian Science Foundation, which co-funded the study.